Particle-Particle Interactions
All particles have a van der Waals attraction brought about by
a variety of forces, principal among these the interaction of
dipoles of neighboring atoms and molecules. This attractive force
is modelled (the Lennard-Jones model) with a r![]() decay
with separation:
decay
with separation:
Eq 33-2:![]()
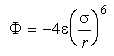
where ![]() is a scaling factor for the energy of interaction
and
is a scaling factor for the energy of interaction
and ![]() is a scaling length for the molecular size. To
a good approximation the force exerted by individual atoms or
molecules is additive so that the force exerted by a particle
can be obtained by integration and the force exerted by a spherical
particle can be described by a constant Q:
is a scaling length for the molecular size. To
a good approximation the force exerted by individual atoms or
molecules is additive so that the force exerted by a particle
can be obtained by integration and the force exerted by a spherical
particle can be described by a constant Q:
Eq 33-3:![]()
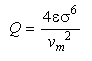
where v![]() is the molecular volume. Q ranges from a few
kT for strongly polar molecules to of order 0.1 kT for crystalline
silicates and oxides. For interaction of two spherical particles
of radius r
is the molecular volume. Q ranges from a few
kT for strongly polar molecules to of order 0.1 kT for crystalline
silicates and oxides. For interaction of two spherical particles
of radius r![]() and r
and r![]() , with centers separated a distance
d the energy of interaction is:
, with centers separated a distance
d the energy of interaction is:
Eq 33-4:![]()
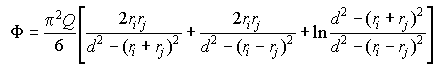
When the particles are much larger than their distance of separation, the energy of interaction decays by approximately the square of the distance between particle edges at closest approach, as marked in red:
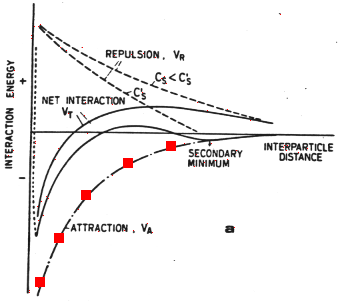
Figure 33-1a
At very close approach the Born repulsion of adjacent electron clouds (marked in blue) begins to play a role, so in the absence of any other forces there is a strong energy minimum with distances of approach of order 1 nm.
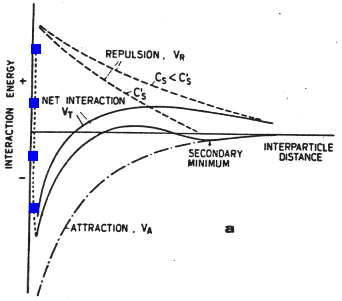
Figure 33-1b
There is an additional important term, the electrostatic interaction of particles. Particles in water generally have a surface charge, acquired by ionization of atoms at the surface of the particle. Here Si represents any cation at the surface of a crystalline oxide or aluminosilicate while the carboxyl group is representative of a functional group that would be found at the surface of particulate organic material (POM). These surfaces exhibit acid-base behavior with surfaces becoming more negatively charged with increasing pH.
Eq 33-5:![]()
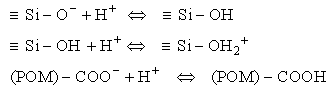
This acid-base behavior leads to positive surface charge at low pH and negative surface charge at intermediate and high pH for a variety of materials. At moderate pH most surfaces are negatively charged.
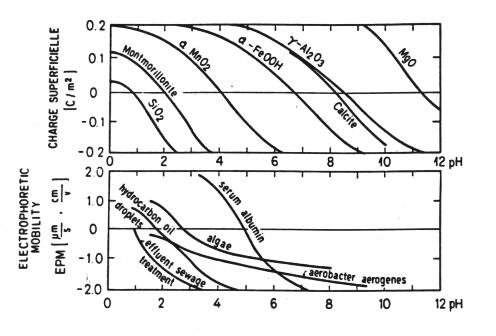
Figure 33-2
How do these charged surfaces interact? First we consider the nature of an ionic fluid near a charged surface. The simplest model aligns an excess of positive ions along a negatively charged surface, generating a potential across a thin layer (the capacitor or Helmholtz model of a surface). However experiment shows this model to be too simple. A more realistic model is that proposed by Guoy and independently by Chapman in the early 1910s. This model holds that ions redistribute themselves in solution to accomodate the gradient of potential brought about by a charged surface
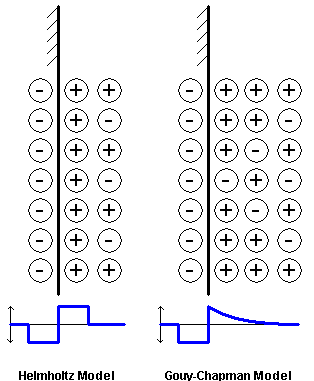
Figure 33-3
At all points the chemical potential:
Eq 33-6:![]()
![]()
is a constant. For positively charged ions:
Eq 33-7:![]()
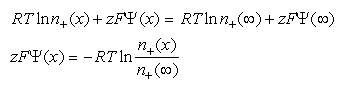
and for negatively charged ions:
Eq 33-8:![]()
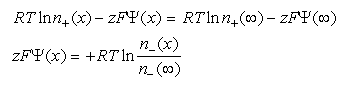
The charge density at any point is:
Eq 33-9:![]()
![]()
Potential and charge q are related by the Poisson equation:
Eq 33-10:![]()
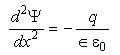
where the product in the denominator on the right hand side is related to the dielectric characteristics of the fluid. This differential equation can be solved subject to appropriate boundary conditions to find that (for low potentials):
Eq 33-11:![]()
![]()
where
Eq 33-12:![]()
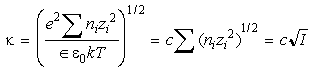
and I is the ionic strength. This characteristic length then determines how the fields of two adjacent particles interact. For aqueous solutions:
Eq 33-13:![]()
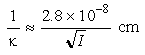
so that in seawater (ionic strength 0.7) the scale length is 0.3
nm and in river water with ionic strength of 10![]() the scale length
is 9 nm.
the scale length
is 9 nm.
The electrostatic repulsion (marked in cyan) and the van der Waals attraction combine to give the overall energy of interaction:
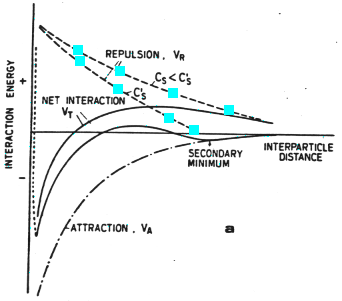
Figure 33-1c
As a function of ionic strength, the overall interaction undergoes a sharp transition from repulsive to attractive:
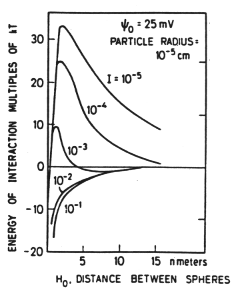
Figure 33-4
This is manifested in the behavior of particles, e.g., as shown in these data from experiments in which suspensions of the clay mineral kaolinite were monitored at different ionic strengths.
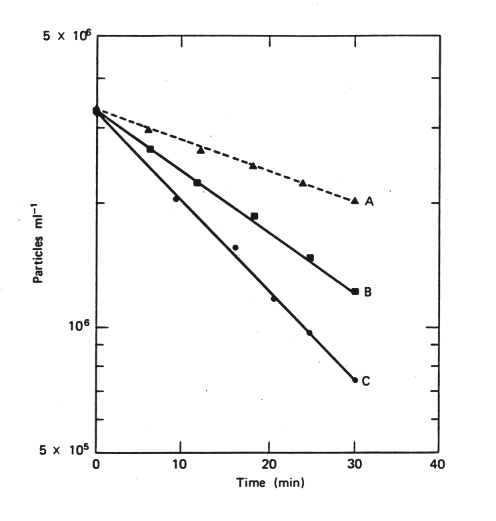
Figure 33-5. A, ionic strength .036; B, ionic strength .087; C, ionic strength .343. From (37)
We have emphasized the tradeoff of repulsive electrostatic forces with van der Waals attraction. There are other way of promoting adhesion between particles. In particular coatings of biopolymers, in addition to having electrostatic interactions, can bridge between particles by forming chemical bonds with a range of energies (hydrogen bonding, covalent bonding).
Particle Collisions
Particles move simply because of their thermal energy, i.e., they undergo Brownian motion. Consider a disperse suspension of one size of particles that have some tendency to aggregate. The rate of change of the number density of particles will be described by:
Eq 33-7:![]()
![]()
where
Eq 33-8:![]()
![]()
In this expression n is the volume density of particles,
t is time, ![]() is the fraction of collisions leading
to aggregation (=1 all adhere, =0 none adhere), D is the thermal
diffusion coefficient of the particles (which is kT/(6
is the fraction of collisions leading
to aggregation (=1 all adhere, =0 none adhere), D is the thermal
diffusion coefficient of the particles (which is kT/(6![]() µ
r)), and r is the particle radius. Simplifying:
µ
r)), and r is the particle radius. Simplifying:
Eq 33-9:![]()
![]()
Eq 33-10:![]()
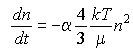
At 25°C, the value of k![]() is 5.5 x 10
is 5.5 x 10![]()
![]() cm
cm![]() s
s![]() .
.
Suppose ![]() is one and consider as an example 1 µm particles
in a 1 g/l suspension. Each particle has a mass of about 10
is one and consider as an example 1 µm particles
in a 1 g/l suspension. Each particle has a mass of about 10![]()
![]() g so the volume density of particles is about 10
g so the volume density of particles is about 10![]() cm
cm![]() . What
is the time to halve the density of particles? Solving
equation 24-5 for the initial condition n(t=0)=n
. What
is the time to halve the density of particles? Solving
equation 24-5 for the initial condition n(t=0)=n![]() :
:
Eq 33-11:![]()
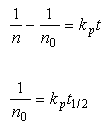
At this particle density, the characteristic time is about 30 minutes. If the particles were a factor of ten larger (10 µm), then the characteristic time would increase to about 21 days. If we compare to the Stokes settling times, we see that the effect is only important for the smallest particles.
The discussion can be made more general by introducing the concept of a coagulation kernel which unifies the treatment of different physical mechanisms for bringing particles together:
Eq 33-12:![]()
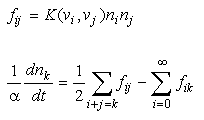
In these expressions, f is the frequency of collision of particles. The subscripts i and j denote size classes of particles of volume v. The time rate of change of particle concentration in the k size class is given by two terms. The first represents the particular combinations of smaller particles that can aggregate to form a particle exactly in size class k. The second represents the disappearance of particles from the k size class due to collision and aggregation with all size classes. The frequency of collision is the product of a coagulation kernel and the particle density in two size classes, e.g., for Brownian motion:
Eq 33-13:![]()
![]()
where the kernel is given by:
Eq 33-14:![]()
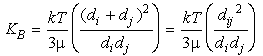
We can compare this expression to our earlier discussion of Brownian motion if we consider a monodisperse (single particle size) suspension:
Eq 33-15:![]()
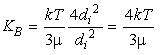
(For a monodisperse suspension there are no smaller particles from which to make particles of the initial size so that the first term in equation 24-7 is zero.)
A useful way of visualizing the magnitude of the kernel is to consider it size for a particular particle size as a function of the size of the other particle:
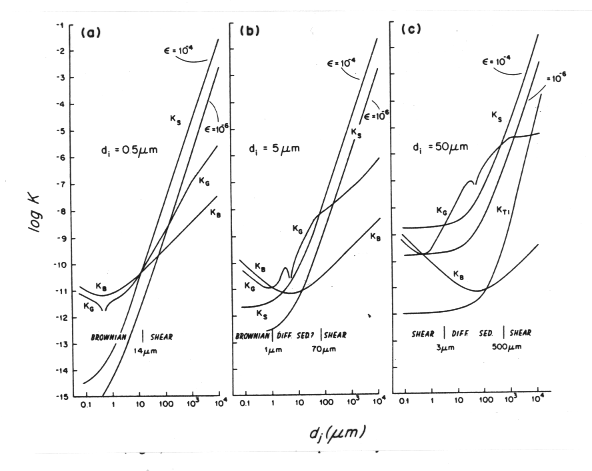
Figure 33-2, (35)
This figure shows (among other things) the Brownian motion
kernel for three different
values of d![]() .
There is a minimum at the point d
.
There is a minimum at the point d![]() =d
=d![]() with a 1/d
with a 1/d![]() dependence
at smaller d
dependence
at smaller d![]() and a d
and a d![]() dependence at larger d
dependence at larger d![]() .
.
Brownian motion is not the only mechanism which can bring particles together. In the presence of shear, particles at one level are traveling at a velocity different from those on another level leading to collisions.
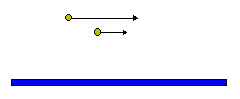
The coagulation kernel due to shear is given by:
Eq 33-16:![]()
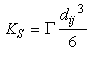
If the flow is laminar:
Eq 33-17:![]()
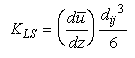
If the flow is turbulent:
Eq 33-18:![]()
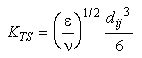
where ![]() is the turbulent dissipation rate and
is the turbulent dissipation rate and ![]() the kinematic viscosity.
the kinematic viscosity.
We can compare the relative sizes of the kernels for Brownian motion and shear:
Eq 33-19:![]()
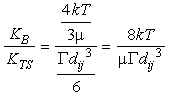
In the lower 100 m of the water column, ![]() ~0.1 s
~0.1 s![]() , so for a monodisperse
suspension the boundary between dominance of the two mechanisms
falls at a particle size given by:
, so for a monodisperse
suspension the boundary between dominance of the two mechanisms
falls at a particle size given by:
Eq 33-20:![]()
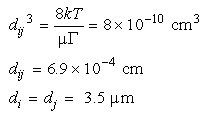
If the suspension is monodisperse, then Brownian motion is more important for particles smaller than 3.5 µm and shear more important is bringing about collisions of particles larger than 3.5 µm.
Figure 33-2 also shows the behavior of the kernel for
collisions due to shear for two different values of the turbulent
dissipation rate. When d![]() is small, the value of the kernel
approaches a constant value controlled by d
is small, the value of the kernel
approaches a constant value controlled by d![]() :
K
:
K![]() ~
~![]() d
d![]() /6. When d
/6. When d![]() is large, the value of the kernel
follows the cubic dependence on d
is large, the value of the kernel
follows the cubic dependence on d![]() : K
: K![]() ~
~![]() d
d![]()
![]() /6.
/6.
Particles also come together by differential settling with larger particles sweeping out smaller particles.
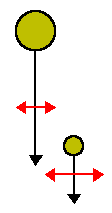
The kernel for this process is given by:
Eq 33-21:![]()
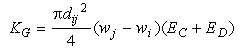
where w is the settling velocity (denoted by the black arrows), E![]() is the collision
cross-section, and E
is the collision
cross-section, and E![]() is the enhancement of the collision
cross
section by Brownian motion (denoted by the red arrows). E
is the enhancement of the collision
cross
section by Brownian motion (denoted by the red arrows). E![]() and
E
and
E![]() are complicated functions of the sizes of the particles and thus
so is the kernel (Figure 33-2).
are complicated functions of the sizes of the particles and thus
so is the kernel (Figure 33-2).
The relative sizes of the coagulation kernels summarized in Figure 33-2 indicates that different mechanisms are important for different sized particles. For very small particles, Brownian motion and shear are important mechanisms. For intermediate sized particles, all three mechanisms can play a role. For the large particles, differential settling is dominant except for interactions with much larger and smaller particles.
It is important to keep in mind that Figure 33-2 is somewhat misleading in that it contains no information on particle size distribution, while the frequency of collisions depend both on the magnitude of the particle densities and the kernel. n(d) is strongly size dependent:
Figure 33-3, (35)
By combining the information in these two figures, we can estimate the residence time of particles in various size classes and compare these to the residence time due to Stokes settling through the lower 100 meters of the water column (these residence times are in years):

Coagulation through Brownian motion moves particles to larger size classes where they are removed by Stokes settling. Note that there is likely to be a maximum in the residence time for particles somewhere between 0.5 and 5 µm.
For more information on this topic see references (35), (36) and (37).
|
| Oceanography 540 Pages Pages Maintained by Russ McDuff (mcduff@ocean.washington.edu) Copyright (©) 1994-2001 Russell E. McDuff and G. Ross Heath; Copyright Notice Content Last Modified 1/2/2001 | Page Last Built 1/2/2001 |